
EU vs. US medical device regulations and quality management: Understanding the key differences
This blog post delves into the primary distinctions between the EU and US approaches to Quality Management Systems (QMS) in the medical device industry.
The world is global—this much is true. However, not all things are equal, even if they may appear to be at first glance. Consider, for example, the topic of QMS within the medical device sector. What are the main differences between the EU and the US?
EU MDR
In the EU, regulations are structured in a way that the actual regulatory text is somewhat general, while the Harmonized Standards provide detailed requirements. Manufacturers must present objective evidence that they fulfill the criteria outlined in both the standards and regulations. The harmonized QMS standard within the EU MDR is ISO 13485, titled ”Medical devices — Quality management systems — Requirements for regulatory purposes”.
US FDA
The US FDA recognizes certain standards as consensus standards, which manufacturers can utilize to demonstrate compliance. ISO 13485 is one such standard. However, while a recognized standard can facilitate the demonstration of compliance with FDA regulations, it does not automatically ensure regulatory approval. The FDA independently evaluates each product to ascertain whether it complies with the agency’s regulations for safety and efficacy.
The quality management system requirements are defined in the regulation “21 CFR Part 820 – Quality System Regulation”. To clarify, this regulation belongs to the United States Code of Federal Regulations (CFR). The CFR is a collection of regulations issued by various federal agencies of the U.S. government, including the Food and Drug Administration (FDA).
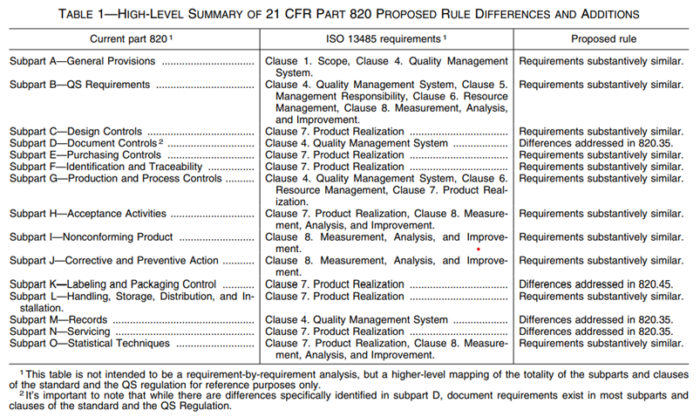
The good news is that the US FDA is integrating ISO 13485:2016 into its regulations. However, some differences will remain, as indicated in Figure 1. On January 31, 2024, the FDA issued the final rule amending the device current good manufacturing practice requirements of the Quality System Regulation under 21 CFR 820.
It is crucial to note that manufacturers with an ISO 13485 certification and/or CE mark will not be exempt from FDA inspections. However, having a QMS based on ISO 13485 can significantly streamline the process of meeting FDA regulatory requirements.
Conclusion
As indicated in the table, there are differences, but broadly speaking, the requirements of US and EU regulations are quite similar. It is important to note ISO 13485 and FDA 21 CFR 820 both aim to ensure the quality, safety, and effectiveness of medical devices. Manufacturers seeking compliance with both ISO 13485 and FDA regulations typically must implement systems that satisfy the distinct requirements of each standard while maintaining overall regulatory compliance.
The advantage for medical device manufacturers is that they do not need to completely rewrite their QMS when transitioning between markets. The downside, however, is that compliance with one set of regulations does not automatically equate to compliance with the other. The devil is in the details, and manufacturers need to understand the requirements and implement the processes wisely.
In this blog we touched the Quality Management part only. There are many more aspects that need to be addressed to achieve full regulatory compliance for the medical device markets.
Useful links:
- EU Harmonized Standards for Medical Devices
- FDA 21 CFR 820
- FDA 21 CFR 820 Guidance
- List of Notified Bodies
- International Medical Device Regulators Forum

Juhani Perhonen
Chief Business Developer
juhani.perhonen@atostek.com
+358 45 7834 5589, +1 914 530 4069 (USA)